Mai 26, 2021
Medizinprodukterecht
Zäsur für die Medizinprodukte-Industrie in Europa
Der Bundesverband Medizintechnologie, BVMed, fordert zum Geltungsbeginn der neuen EU-Medizinprodukte-Verordnung (MDR) am 26. Mai 2021 schnelle und pragmatische Lösungen und eine bessere Unterstützung der kleinen und mittelständischen Unternehmen. „Wir müssen Benannte Stellen schneller notifizieren, Remote Audits zulassen, die Übergangsfrist für Altzertifikate verlängern sowie pragmatische Lösungen für bewährte Bestandsprodukte und Nischenprodukte etablieren“, so BVMed-Vorstandsvorsitzender Dr. Meinrad Lugan und Vorstandsvize Marc D. Michel. Insgesamt hält der BVMed die MDR für noch nicht praxistauglich. Es gebe zu wenige Benannte Stellen, zu bürokratische Vorschriften und fehlende Klarstellungen. „Wir müssen Lösungen finden, die MDR strategisch weiterentwickeln und den MedTech-Standort stärken, denn ein Stillstand hätte fatale Folgen für die Patientenversorgung und für den mittelständisch geprägten Medizintechnik-Standort Deutschland“, so der deutsche Medizintechnik-Verband.
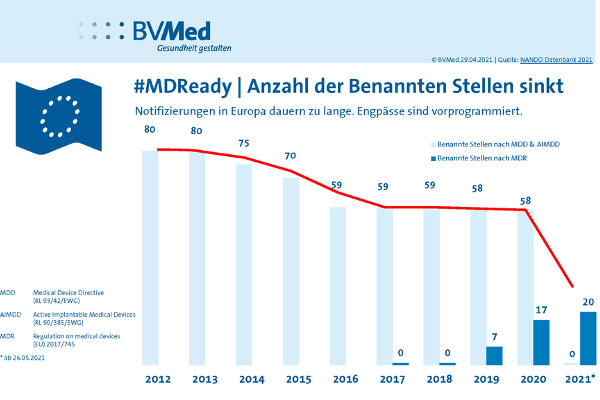
Bild: BVMed e.V.
Am 26. Mai 2021 ist Geltungsbeginn der EU-Medizinprodukte-Verordnung (Medical Device Regulation – MDR). Dieses Datum ist eine Zäsur für die Medizinprodukte-Industrie in Deutschland und Europa, weil die Anforderungen an den Marktzugang von Medizinprodukten, an den Lebenszyklus des Produkts und an die Benannten Stellen erheblich steigen. Es handelt sich um einen völlig neuen Rechtsrahmen – eine Revolution, keine Evolution. Ziel der MDR ist es, die Patientenversorgung mit sicheren Medizinprodukten zu gewährleisten und die Patientensicherheit zu erhöhen. „Dieses Ziel teilt die MedTech-Branche voll und ganz“, so der BVMed.
Erhöhte Anforderungen und bürokratischer Aufwand
Die MDR bringt mit den erhöhten Anforderungen einen um gut das Zehnfach gestiegenen bürokratischen Aufwand und dramatische Kostensteigerungen mit sich, die vor allem die kleinen und mittelständischen Medizinprodukte-Unternehmen bedrohen.
- Die deutlich höheren Anforderungen beziehen sich beispielsweise auf die Generierung klinischer Daten sowie die Nachverfolgung des Medizinproduktes über dessen gesamten Lebenszyklus.
- Der neue Rechtsrahmen betrifft dabei nicht nur neue Medizinprodukte, sondern auch alle bewährten Bestandsprodukte.
- Zudem müssen alle Benannten Stellen einen langwierigen europäischen und multinationalen Benennungsprozess durchlaufen, da die bisherigen Benennungen mit Geltungsbeginn der MDR ihre Gültigkeit verlieren.
- Die Benannten Stellen brauchen deutlich mehr Prüfzeit für die umfangreicheren Akten. Ein Wechsel der Benannten Stelle ist aufgrund der aktuellen Engpässe praktisch ausgeschlossen.
Offene MDR-Baustellen
Die einjährige Verschiebung des Geltungsbeginns hätte vor allem den Behörden Zeit verschafft, das System regulatorisch arbeitsfähig zu machen. Dies ist jedoch nicht hinreichend genutzt worden. Die Unternehmen setzen sich für hohe Qualitätsstandards ein und leisten ihren Beitrag für eine bestmögliche Patientenversorgung. Das muss in einem Regelwerk definiert sein. Das Regelwerk muss dann aber auch umsetzbar sein. Das ist bei der MDR noch immer nicht der Fall. Die offenen Baustellen in der Übersicht:
- Anzahl und Kapazitäten der unter der MDR Benannten Stellen sind noch immer viel zu gering, um alle Bestandsprodukte fristgerecht in die MDR zu überführen. Aktuell sind es erst 20 Benannte Stellen. Unter dem alten Recht waren es zuletzt mehr als 50. Viele Hersteller – zum Beispiel von chirurgischen Instrumenten oder Software – müssen Neuverträge mit Benannten Stellen abschließen. Das ist praktisch unmöglich, weil es keine ausreichenden Kapazitäten gibt und vorerst vornehmlich Bestandskunden bedient werden. Das gleiche Problem haben Start-ups im MedTech-Bereich.
- Die Übergangsfrist für die Gültigkeit der Altzertifikate („Grace Period“) bis Mai 2024 und die Abverkaufsfrist bis Mai 2025 wurden bei der Verschiebung des Geltungsbeginns nicht angepasst – trotz der Auswirkungen der Corona-Pandemie. Die zu erwartenden Engpässe bleiben also bestehen und haben sich sogar noch verstärkt.
- Die rund 20.000 Zertifikate werden so bis zum Ende der Übergangsperiode im Mai 2024 nicht in die MDR überführt werden können. Aktuell sind erst rund 180 MDR-Zertifikate ausgestellt. Die zehnfache Menge davon liegt den Benannten Stellen als Antrag vor. Es ist abzusehen, dass sich die Lage 2024 dramatisch zuspitzen wird. Viele Medizinprodukte werden es nicht rechtzeitig in die MDR schaffen. Es drohen Versorgungsengpässe, Innovationsstau und Wettbewerbsverzerrung.
- Die Zertifizierungsprozesse werden seit über einem Jahr immer noch durch Reise- und Quarantänebeschränkungen massiv verzögert. Audits bei Herstellern finden nur teilweise statt und können nicht abgeschlossen werden. Für Remote Audits ist aber auch nach über einem Jahr Corona-Pandemie keine ausreichende rechtliche Basis vorhanden. Das Vorgehen der Mitgliedsstaaten ist nicht harmonisiert. Während Frankreich, Irland oder die Niederlande pragmatisch vorgehen, sind Deutschland und Italien restriktiv. Das Ergebnis ist eine Wettbewerbsverzerrung im europäischen und internationalen Markt.
- Für Bestandsprodukte sind klinische Daten im Sinne der MDR-Anforderungen nicht immer ausreichend vorhanden. Für derartige Untersuchungen und Studien bei lang bewährten Produkten findet man aber weder Probanden, noch Prüfärzte. Hinzu kommt, dass positive Voten von Ethikkommissionen schwer zu erhalten sind, da klinische Prüfungen mit langjährigen Bestandsprodukten ethisch nicht vertretbar sind.
- Für Orphan Devices, also Nischenprodukte, fehlen Ausnahmeverfahren.
- Die Datenbank EUDAMED, das digitale Rückgrat der MDR, ist noch nicht funktionsfähig.
- MDCG-Leitlinien, die als Implementierungshilfe gedacht sind und die MDR für eine harmonisierte Umsetzung interpretieren sollen, sind teilweise uneinheitlich, verkomplizieren die Anforderungen oder gehen darüber hinaus und werden ohne jegliche Übergangsfristen erstellt.
- Innovationen stecken aufgrund der MDR-Bürokratie in der Warteschleife und drohen auszuwandern. Wenn 50 Prozent der F&E-Ausgaben in Re-Zertifizierungen von Altprodukten gehen, dann läuft etwas falsch. Darunter leidet der medizintechnische Fortschritt. Wir verschenken damit auch Wertschöpfung in Deutschland.
BVMed-Vorstandsvorsitzender Dr. Meinrad Lugan: „Wenn insbesondere kleine und mittelständische Firmen gezwungen sind, alle ihre Entwicklungsressourcen in die Regulatorik zu verlagern, und zwar auf Kosten der Innovationstätigkeit, dann zeigt das, dass man mit der MDR augenscheinlich etwas über das Ziel hinausgeschossen ist. Hersteller geraten zunehmend unter Druck. Zertifizierungsverfahren laufen nicht an, verzögern sich, weil die Re-Zertifizierungen der Bestandsprodukte die Kapazitäten binden.“
Die Branche braucht schnelle und pragmatische Lösungen
Mit Blick auf die Baustellen muss festgestellt werden, dass das MDR-Regelwerk aktuell nicht praxistauglich ist. Wir müssen auf die aktuelle Situation reagieren und Lösungen finden. Ein Stillstand hat fatale Folgen für die Patientenversorgung und für den Medizintechnik-Standort Deutschland.
- Benannte Stellen müssen in einer konzertierten Aktion aller beteiligten Behörden schneller notifiziert werden. Alle Scopes (Fachspektren) müssen ausreichend abgedeckt sein. Es müssen genügend Ressourcen in den Benannten Stellen vorhanden sein. Für Hersteller, die nachweislich keine Benannte Stelle finden, müssen Lösungen etabliert werden.
- Die Übergangsphase und die Laufzeit der Zertifikate müssen verlängert werden, um den abzusehenden Engpass im Jahr 2024 zu entzerren.
- Für Remote Audits muss die Europäische Kommission schnellstmöglich eine rechtliche Basis schaffen.
- Für bewährte Bestandsprodukte müssen pragmatische Lösungen beispielsweise über das Instrument der „Anerkennung klinischer Praxis“ gefunden werden.
- Für „Orphan Devices“ (Nischenprodukte) muss die Europäische Kommission Ausnahmeregelungen nach dem US-Vorbild der „Humanitarian Device Exemption“ sowie der „Orphan Drug“-Regelungen in Europa schaffen.
- Für KMU sollten spezielle Förderprogramme beispielsweise zur Unterstützung von klinischen Studien aufgelegt werden. Diese Förderprogramme dürfen sich nicht nur auf Neuentwicklungen und Innovationen beschränken, sondern müssen Bestandsprodukte einschließen.
- Für die Marktbeobachtung benötigen wir ein agiles und digitales „Post-Market-Surveillance“-System, um die Patientensicherheit weiterhin zu gewährleisten. Dabei muss das gesamte Gesundheitssystem eingebunden werden: Fachgesellschaften, Krankenhäuser, Krankenkassen. Es müssen DSGVO-konforme Möglichkeiten geschaffen werden, damit die Unternehmen klinische Daten nutzen können. Das Implantate-Register-Gesetz ist dafür ein Anfang.
BVMed-Vorstandsvorsitzender Dr. Meinrad Lugan und BVMed-Vize Marc D. Michel: „Wir brauchen pragmatische Lösungen und regulatorische Korrekturen. Unser europäisches System muss Geschwindigkeit aufnehmen, bürokratische Hürden abbauen und zeitliche und finanzielle Berechenbarkeit für seine Medizinprodukte-Industrie bieten. Deshalb müssen wir die MDR rechtzeitig und vor allem strategisch weiterentwickeln, damit der medizintechnische Fortschritt in Europa auch in Zukunft beim Patienten ankommt und wir unsere mittelständisch geprägte Medizintechnik-Branche stärken.“
Den aktuellen Umsetzungsstand beleuchtet die BVMed-Akademie auf einer MDR-Branchenkonferenz am 26. Mai 2021 unter anderem mit Bundesgesundheitsminister Jens Spahn.
Quelle: BVMed e.V.